Article
An international collaboration of researchers tested the strength of protein complexes and the results could provide insights that can help develop strategies to prevent bacteria resistance.
Bacterial cell walls have several surface proteins that help them degrade different materials, including plant fibers. These protein complexes, named cellulosomes, are linked together by very strong interactions.
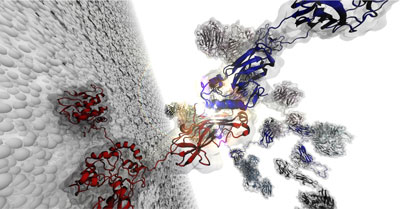
“We are interested in proteins that are very resilient to force,” said Rafael Bernardi, a research scientist at the Beckman Institute. Bernardi and his colleagues studied a protein system that can withstand up to 1 nN. According to Hermann Gaub, a professor at Ludwig Maximilian University of Munich and an author on the paper, the system is about 1000 times stronger than the muscle motor protein that allows us to move our arms and legs.
“The whole system is a very large complex of proteins,” Bernardi said. “We were interested in using computational methods to understand how they assemble because they are like Lego bricks: You have a lot of them and you can assemble them in different ways.”
After making the computational models, Bernardi’s collaborators in Munich and Basel, make the protein complexes in the lab and try to pull them apart to understand how the complexes are assembled. “When they started doing the experiments, we realized that these complexes are much stronger than most systems that have been studied before,” Bernardi said.
The results are detailed in the paper “Mechanisms of Nanonewton Mechanostability in a Protein Complex Revealed by Molecular Dynamics Simulations and Single-Molecule Force Spectroscopy” that was published in the Journal of the American Chemical Society.
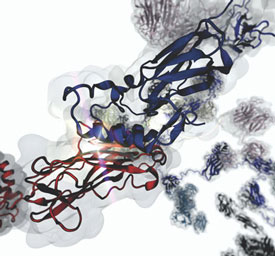
Protein systems that can withstand forces are important for several reasons. “There are bacteria that can adhere to implants and cause infections,” said Zaida “Zan” Luthey-Schulten, a professor of biophysics and quantitative biology and a Murchison-Mallory Endowed Chair in Chemistry. “With these insights, we can come up with strategies to prevent that.”
Bernardi uses the Blue Waters supercomputer at the University of Illinois to make his computational models. “We know the position of every atom in the protein at every moment in time,” Bernardi said. “We can then use intelligent design to tell the experimentalists what aspects are important. All our simulations were able to predict what they found in the experiment.”
The computational models work the best when there are strong forces involved. In the future, Bernardi is interested in looking at proteins that are involved with weaker forces. “There is a lot of biology that involves weaker forces, for example proteins like myosin. If you make a single mutation that increases the force resilience, you can cause heart problems,” Bernardi said.
“We really like integrating experiments, theory, and simulations,” Luthey-Schulten said. “It’s nice when you can start making predictions and you have willing collaborators who will do the experiments. Without Bernardi’s simulations, Gaub’s group would not know which experiments to do next.”
The paper, “Mechanisms of Nanonewton Mechanostability in a Protein Complex Revealed by Molecular Dynamics Simulations and Single-Molecule Force Spectroscopy,” can be found at https://doi.org/10.1021/jacs.9b06776
Beckman Institute for Advanced Science and Technology